Bài viết Tìm hiểu về các kiến thức liên quan đến di truyền học của sẹo và sẹo lồi – Tải file PDF Tại đây.
Tác giả: Chao-Kai Hsu, Hsing-San Yang và John A. McGrath
Dịch: Bs. Trương Tấn Minh Vũ
Sẹo lồi (keloid) là tổn thương lớp bì nhô lên do quá trình lành thương bất thường. Không giống như sẹo phì đại, trong đó tổn thương lớp bì nhô lên nằm trong giới hạn của vết thương ban đầu, sẹo lồi phát triển vượt ra ngoài ranh giới vết thương ban đầu. Về mặt mô bệnh học, sẹo lồi đặc trưng bởi collagen sẹo lồi (sợi collagen dày ưa eosin hyalin hóa) với rìa tiến triển ngang dạng lưỡi ở lớp bì. Sẹo lồi thường bắt đầu sau nhiều tháng, có thể lên đến 1 năm, sau chấn thương da hoặc quá trình viêm, như mụn trứng cá, viêm nang lông, thủy đậu hoặc tiêm vaccin. Quá trình lành thương bình thường liên quan đến sự biểu hiện của nhiều gen và các con đường truyền tín hiệu phức tạp, trong đó sự tương tác của các gen, phân tử và con đường này có nghĩa là bất kỳ thay đổi nào trong biểu hiện gen đều có thể dẫn đến phản ứng lành thương bất thường, bao gồm cả sự hình thành sẹo lồi. Cơ chế sinh bệnh của sẹo lồi rất phức tạp. Dữ liệu liên quan đến cả các yếu tố tại chỗ trong da (sức căng mô, cytokine và các yếu tố tăng trưởng/thụ thể và các con đường truyền tín hiệu của chúng) và khuynh hướng di truyền, bao gồm các ảnh hưởng biểu sinh. Tóm lại, các cơ chế được đề xuất của sự hình thành sẹo lồi bao gồm: (1) tác động cơ học, (2) tính nhạy cảm về mặt di truyền, (3) rối loạn điều hòa của nhiều cytokine/yếu tố tăng trưởng và (4) sự thay thế collagen bất thường. Tuy nhiên, không có giả thuyết thống nhất nào có thể giải thích đầy đủ về sự hình thành sẹo lồi. Trong chương này, chúng tôi tập trung vào các đóng góp di truyền vào quá trình bệnh sinh sẹo lồi.
4.1 Bằng chứng/ hiện tượng về cơ sở di truyền của sẹo lồi
Có một lượng lớn bằng chứng hoặc hiện tượng cho thấy có yếu tố di truyền đối với sẹo lồi. Về cơ bản, sự khác biệt về tỷ lệ mắc sẹo lồi giữa các quần thể có tổ tiên khác nhau cung cấp manh mối đầu tiên cho thấy khả năng bị sẹo lồi có thể di truyền. Mặc dù sẹo lồi có thể xuất hiện ở mọi quần thể, nhưng người ta đều biết rằng các dân tộc có sắc tố da tối màu, như người Châu Á và Châu Phi, dễ mắc sẹo lồi hơn (lên đến 15 lần). Điều này cũng được phản ánh trong tỷ lệ mắc sẹo lồi là vấn đề da phổ biến thứ năm ở bệnh nhân da đen trưởng thành tại Vương quốc Anh.
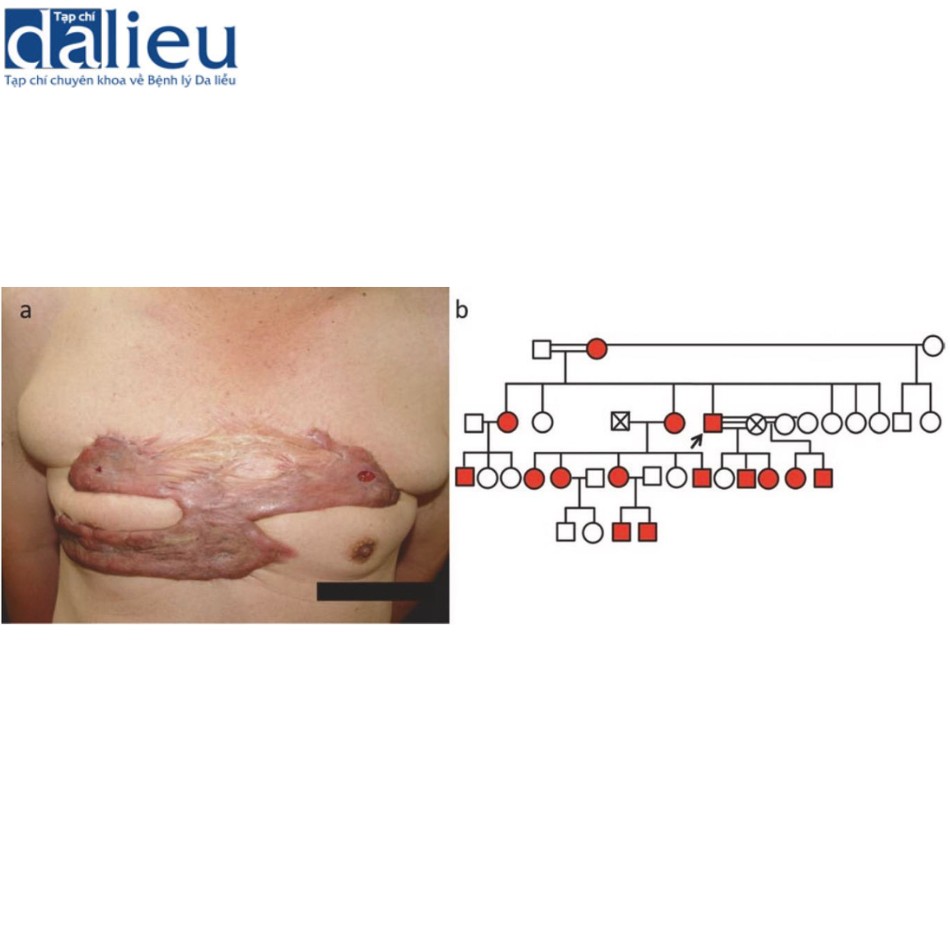
Thứ hai, có nhiều nghiên cứu được tiến hành về “phả hệ sẹo lồi”, trong đó khả năng mắc sẹo lồi được chứng minh là di truyền trong các dòng dõi gia đình. Omodare và cộng sự lần đầu tiên báo cáo về sẹo lồi có kiểu di truyền lặn trên nhiễm sắc thể thường trong 34 gia đình từ một quần thể người Nigeria. Marneros và cộng sự ghi nhận di truyền trội trên nhiễm sắc thể thường với độ thâm nhập lâm sàng không hoàn toàn và biểu hiện thay đổi trong số 14 gia đình sẹo lồi, chủ yếu là người Mỹ gốc Phi (n = 10), nhưng cũng bao gồm người da trắng (n = 1), người Nhật Bản (n = 2) và người Phi-Caribê (n = 1). Kiểu di truyền trội trên nhiễm sắc thể thường được hỗ trợ thêm bởi một số nghiên cứu về người Hán, người gốc Phi-Caribê, người Mỹ gốc Phi và người Mỹ gốc Á. Trong một nhóm 750 bệnh nhân sẹo lồi người Đài Loan, hơn một nửa báo cáo có tiền sử gia đình dương tính, với hầu hết có di truyền trội trên nhiễm sắc thể thường với độ thâm nhập không hoàn toàn (dữ liệu chưa công bố, Hsu CK) (Hình 4.1). Nhìn chung, một chế độ di truyền duy nhất cho kiểu hình sẹo lồi có vẻ không có khả năng xảy ra, nhưng di truyền trội trên nhiễm sắc thể thường với độ thâm nhập không hoàn toàn và biểu hiện thay đổi, dường như có khả năng xảy ra nhất dựa trên các báo cáo từ một số nhóm độc lập trong các nhóm dân tộc khác nhau.
Nghiên cứu về cặp song sinh cũng là công cụ mạnh mẽ để điều tra mức độ mà di truyền góp phần gây ra sẹo lồi. Trong cặp song sinh cùng trứng có chung 100% gen, cặp song sinh khác trứng chỉ có chung trung bình 50%. Không khó để hiểu được tầm quan trọng của yếu tố di truyền trong bệnh sinh sẹo lồi trong khi các bệnh có tỷ lệ phù hợp cao giữa các cặp song sinh cùng trứng
4.2 Các hội chứng liên quan đến sẹo lồi
Một số hội chứng bẩm sinh hiếm gặp được báo cáo là có liên quan đến sẹo lồi có tính gia đình. Các hội chứng này chủ yếu là các rối loạn theo thuyết di truyền của Mendel, có cơ sở phân tử đã được biết, và cung cấp cơ hội tốt để nghiên cứu bệnh sinh của sẹo lồi và tìm manh mối nguyên nhân, phương pháp điều trị khả thi hoặc các biện pháp phòng ngừa.
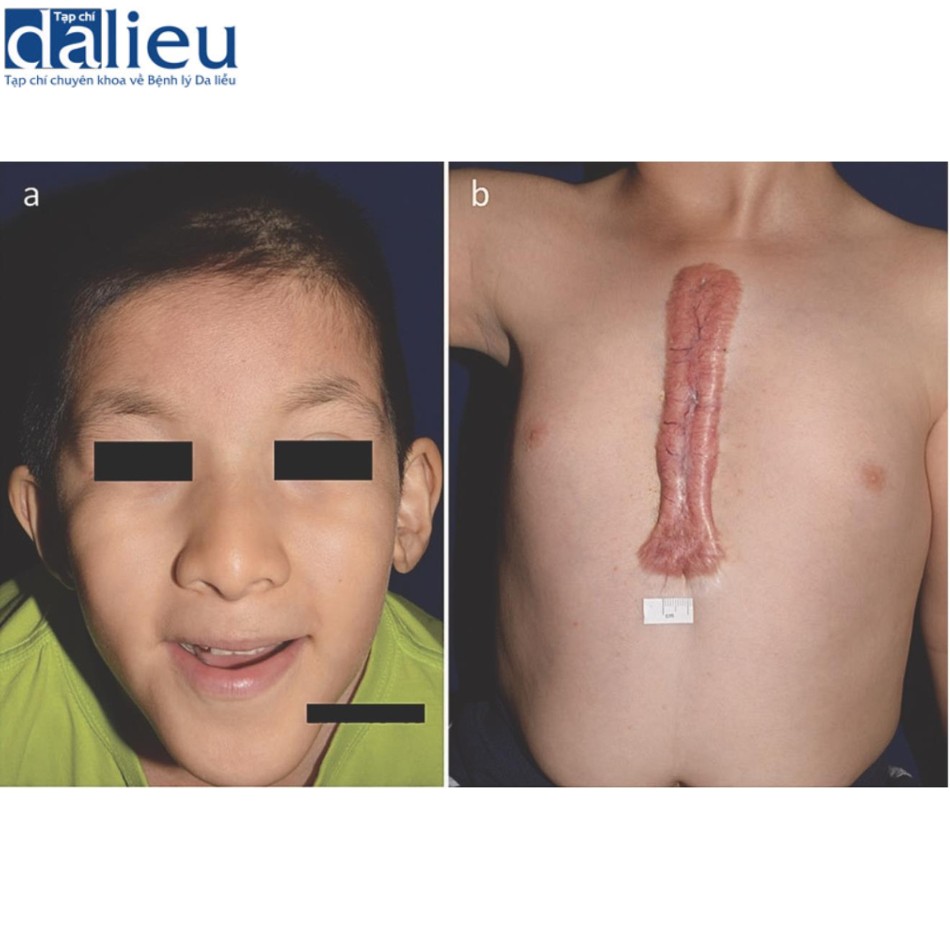
Hội chứng Rubinstein–Taybi (RSTS1; OMIM 180849, RSTS2; 613684) là hội chứng liên quan đến sẹo lồi phổ biến nhất. Rối loạn này được đặc trưng bởi chậm phát triển trí tuệ, chậm phát triển, đầu nhỏ, các đặc điểm khuôn mặt dị dạng. Có một số đặc điểm khuôn mặt riêng biệt, như lông mày cong cao, sống mũi rộng, mũi khoằm, nụ cười nhăn nhó và tỷ lệ sẹo lồi tăng (Hình 4.2). Một nghiên cứu hồi cứu cho thấy 15 trong số 62 cá nhân người Hà Lan mắc RTS bị sẹo lồi, tự phát hoặc sau chấn thương nhẹ, thường bắt đầu trong giai đoạn dậy thì sớm. RTS là do đột biến gen CREBBP hoặc EP300 với di truyền trội trên nhiễm sắc thể thường. Gen CREBBP mã hóa cho protein liên kết CREB, trong khi EP300 mã hóa cho protein histone acetyltransferase p300. Cả hai đều là đồng hoạt hóa trong các protein liên quan đến SMAD/con đường truyền tín hiệu yếu tố tăng trưởng biến đổi (TGF)-β. Hơn nữa, sự tham gia của histone acetyltransferase chỉ ra biến đổi biểu sinh có thể đóng vai trò trong quá trình bệnh sinh của sẹo lồi, điều này được một số nghiên cứu ủng hộ.
Atwal và cộng sự gần đây báo cáo một hội chứng liên kết X mới của bệnh van tim, giảm khả năng vận động của khớp và sẹo lồi do đột biến ở FLNA (c.4726G>A; p.G1576R). Protein được mã hóa, filamin A, là một protein liên kết với SMAD cung cấp một khung cho sự tương tác giữa TGF-β và các con đường dẫn truyền tín hiệu khác.
Hội chứng Ehlers–Danlos (EDS) là một nhóm các rối loạn mô liên kết di truyền không đồng nhất về mặt lâm sàng và di truyền, đặc trưng bởi tình trạng tăng động của khớp, da tăng độ giãn và mô dễ vỡ. Phân loại năm 2017 mô tả 13 loại EDS. Trong số này, các mảng sẹo lồi ở các chi dưới được báo cáo ở loại EDS IV, nguyên nhân là do đột biến dị hợp tử ở gen collagen loại III (COL3A1).
Nhìn chung, ba hội chứng di truyền theo Mendel khác nhau này cung cấp bằng chứng về sự đóng góp của một gen vào quá trình sinh tổng hợp sẹo lồi, bất chấp bản chất thay đổi của các bệnh lý chính trong những tình trạng đa dạng này.
4.3 Phân tích sự liên kết xác định một số vị trí và gen ứng viên
Đối với những bệnh nhân bị sẹo lồi không liên quan hội chứng, mặc dù yếu tố di truyền trong gia đình đóng vai trò nổi bật trong việc hiểu biết của chúng ta về ảnh hưởng của gen, nhưng việc xác định các gen có khuynh hướng gây bệnh vẫn rất khó khăn. Phân tích liên kết di truyền là một công cụ mạnh để phát hiện vị trí nhiễm sắc thể của các gen có khả năng gây bệnh: dựa trên quan sát rằng các gen nằm gần nhau về mặt vật lý trên nhiễm sắc thể vẫn liên kết trong quá trình giảm phân và do đó là một kỹ thuật phù hợp để nghiên cứu những cá nhân dễ bị sẹo lồi.
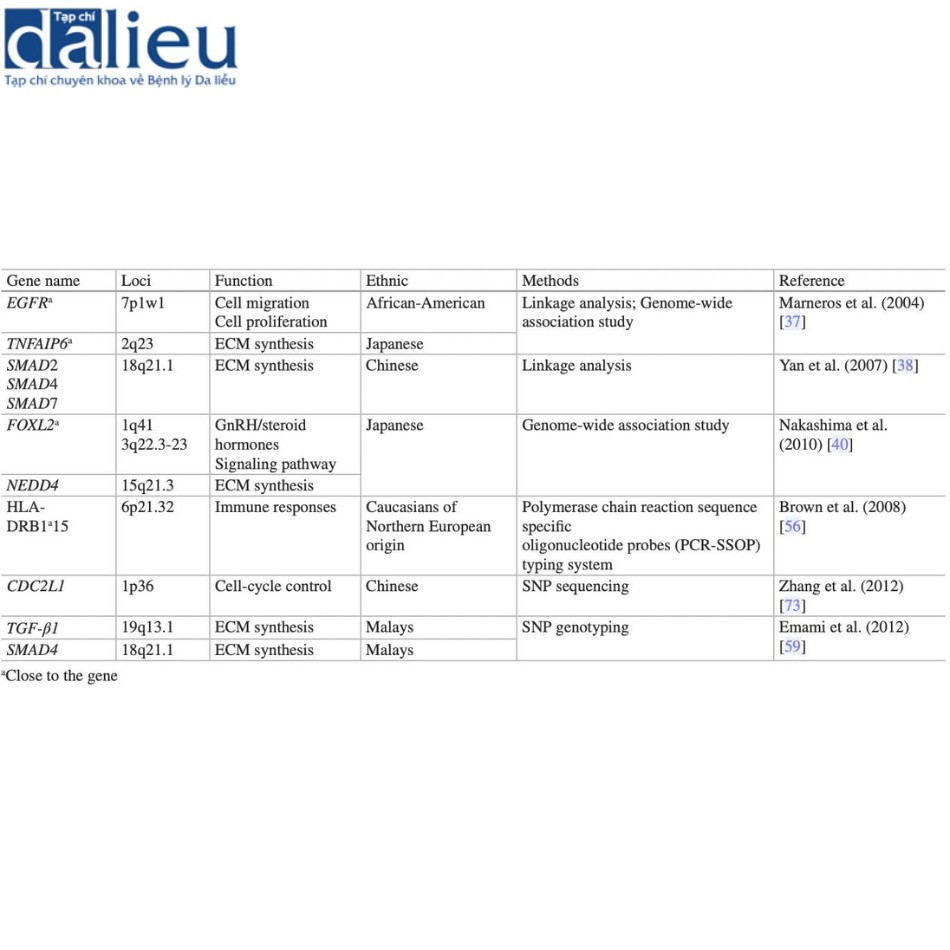
Sử dụng phân tích liên kết trên các gia đình bị sẹo lồi có kiểu di truyền trội trên nhiễm sắc thể thường, các vị trí dễ mắc bệnh được xác định trong một gia đình người Nhật Bản với dải nhiễm sắc thể 2q23 và trong một gia đình người Mỹ gốc Phi với dải nhiễm sắc thể 7p11, cả hai đều có điểm số logarit của tỷ lệ cược (LOD) lớn hơn 3(Bảng 4.1). Gen TNFAIP6 ở dải nhiễm sắc thể 2q23 và gen EGFR ở dải nhiễm sắc thể 7p11 được đề xuất là gen ứng viên trong các locus tương ứng này. Trong một gia đình người Trung Quốc lớn bị sẹo lồi, các khoảng liên kết được tìm thấy ở 15q22.31-q23, 18q21.1 và 10q23.31. Vùng 18q21.1 chứa các gen SMAD2, SMAD7 và SMAD4, có liên quan đến việc điều hòa các con đường truyền tín hiệu TGFβ. Tuy nhiên, giải trình tự Sanger của các gen cụ thể này trong các phả hệ tương ứng không xác định được các biến thể độc nhất.
4.4 Nghiên cứu sự liên quan toàn bộ hệ gen (GWAS)
Nghiên cứu sự liên kết toàn bộ hệ gen (GWAS) là một nghiên cứu quan sát về một bộ các đa hình nucleotide đơn (SNP) trên toàn bộ hệ gen ở các cá nhân khác nhau để xác định xem bất kỳ SNP nào có liên quan đến bệnh ở người hay không [39]. Đơn giản là hàng triệu biến thể di truyền của mỗi đối tượng trong cả nhóm bệnh và nhóm chứng được đọc bằng dãy SNP. Nếu biến thể (các biến thể) thường xuyên hơn trong nhóm bệnh, thì biến thể (các biến thể) được cho là có liên quan đến bệnh. Sau đó, vùng được đánh dấu bằng các SNP đã xác định được coi là “điểm nóng” của bộ gen người đối với bệnh. Do đó, GWAS là một kỹ thuật kiểm tra có khả năng hữu ích để xác định tính nhạy cảm của bộ gen đối với sẹo lồi ở một số lượng lớn cá nhân bị ảnh hưởng và đối chứng.
Bằng thực hiện nghiên cứu GWAS trên 824 cá nhân bị sẹo lồi và 3205 đối chứng trong một quần thể người Nhật Bản, Nakashima và cộng sự xác định được bốn SNP ở ba vùng nhiễm sắc thể: rs873549 ở 1q41, rs940187 và rs1511412 ở 3q22.3 và rs8032158 ở 15p21.3. Trong số này, rs8032158 được tìm thấy trong intron của gen NEDD4. NEDD4 được biết là có tác dụng tăng cường biểu hiện của fibronectin và collagen loại 1 đóng vai trò trong quá trình tích tụ chất nền ngoại bào. Ngoài ra, NEDD4 tham gia vào quá trình ubiquitin hóa và ổn định của thụ thể yếu tố tăng trưởng giống insulin IGF-1 và tình trạng biểu hiện quá mức của thụ thể IGF-1 được quan sát thấy trong các tổn thương sẹo lồi. Các locus này (1q41 và 15q21.3) được xác nhận thêm ở 714 bệnh nhân Trung Quốc bị sẹo lồi. Ogawa và cộng sự xác định thêm rs8032158 tại 15p21.3 là một dấu ấn sinh học về mức độ nghiêm trọng của sẹo lồi ở 204 bệnh nhân Nhật Bản.
4.5 Các nghiên cứu khác trên toàn bộ hệ gen về sẹo lồi
Lập bản đồ hỗn hợp là một phương pháp lập bản đồ gen mạnh mẽ cho các bệnh hoặc đặc điểm cho thấy nguy cơ khác biệt theo tổ tiên và do đó cũng có thể áp dụng để đánh giá các yếu tố di truyền gây ra sẹo lồi. Lập bản đồ hỗn hợp thường được áp dụng cho người Mỹ gốc Phi có nguồn gốc từ người châu Âu và Tây Phi. Velez Edwards và cộng sự áp dụng lập bản đồ hỗn hợp và xác định một vị trí ở 15q21.2-22.3 liên quan đến sự hình thành sẹo lồi ở 478 người Mỹ gốc Phi. Phân tích sâu hơn cho thấy mối liên quan đáng kể tại MYO1E (myosin 1E) và quét bộ gen cũng xác định sự liên quan tại MYO7A (myosin 7A). Hai gen myosin này cho thấy rằng bộ khung tế bào bị thay đổi có thể góp phần vào các đặc tính di cư và xâm lấn được tăng cường của nguyên bào sợi sẹo lồi.
Shih và Bayat thực hiện lai ghép bộ gen so sánh dựa trên mảng trên mô sẹo lồi so với mô đối chứng bên trong và bên ngoài, và xác định các biến thể số bản sao trong 6p21.32, 11q11, 17q12, 8p23.1, 22q13.1, 19p13.1 và 2q14.3 [53]. Mối liên quan giữa sự hình thành sẹo lồi và HLA-DRB5, nằm ở vùng 6p21.32, được xác nhận thêm bằng nghiên cứu qPCR xác nhận. Các mối liên quan HLA khác được báo cáo, bao gồm HLA-DRB1∗15 ở người da trắng và người Trung Quốc, cũng như HLADQA∗104, DQB1∗0501 và DQB1∗0503, cùng nhau ám chỉ sự đóng góp đáng kể của miễn dịch di truyền vào tình trạng dễ bị sẹo lồi [54–56] . Tuy nhiên, nhìn chung, không có gen độc nhất, tập hợp gen hoặc con đường chức năng liên quan nào có thể trực tiếp liên quan đến các yếu tố dễ mắc bệnh hàng đầu về gen và cần nghiên cứu thêm để xác định xem các gen hoặc SNP có thực sự góp phần vào quá trình hình thành sẹo lồi hay không.
4.6 Nghiên cứu di truyền khác về sẹo lồi
4.6.1 TGF-β và SMAD
Về mặt liên kết gen riêng lẻ, có rất nhiều bằng chứng cho thấy sự rối loạn điều hòa của TGF-β và các phân tử tín hiệu hạ lưu SMAD trong quá trình hình thành sẹo lồi. Do đó, một số nghiên cứu tập trung vào các đa hình trong các gen này, bao gồm TGF-β1, TGF-β2, TGF-β3, thụ thể TGF-β (TGF-βR)I, TGFβRII, TGF-βRIII, SMAD3, SMAD6 và SMAD7, và cách chúng có thể góp phần vào quá trình hình thành sẹo lồi.
Tuy nhiên, một số hạn chế có thể có trong các nghiên cứu di truyền này. Đầu tiên, các nghiên cứu này bao gồm quy mô quần thể nhỏ và thiếu các nhóm đối chứng khỏe mạnh. Thứ hai, các gen được chọn để nghiên cứu đột biến hoặc đa hình không được chọn một cách có hệ thống và do đó có thể khó đưa ra kết luận về cơ sở di truyền của quá trình hình thành sẹo lồi. Tu và cộng sự tiến hành phân tích tổng hợp năm nghiên cứu ca-chứng bao gồm tổng cộng 564 trường hợp sẹo lồi và 620 trường hợp chứng khỏe mạnh. Kết quả cho thấy rằng đa hình TGF-β1 (c.509C/T) không liên quan đến tình trạng dễ bị sẹo lồi. Kết quả âm tính của phân tích này cho thấy có một gen thượng nguồn hoặc các gen khác góp phần vào biểu hiện của gen TGFB trong sẹo lồi. Các nghiên cứu chất lượng cao và quy mô lớn được bảo đảm để xác nhận thêm những kết quả này.
4.6.2 TP53
Chất ức chế khối u p53 được chứng minh là đóng vai trò quan trọng trong việc kiểm soát sự tăng sinh và chết theo chương trình của tế bào, do đó, tình trạng rối loạn điều hòa tiềm ẩn của nó trong quá trình sinh bệnh sẹo lồi được nghiên cứu. Saed và cộng sự báo cáo đột biến gen TP53 trong bảy mẫu mô sẹo lồi nhưng không có đột biến nào trong mô da khỏe mạnh hoặc tăm bông má lấy từ cùng một bệnh nhân. Tuy nhiên, kết quả của họ không được một nhóm khác xác nhận.
4.6.3 ASAH1
Santos-Cortez và cộng sự gần đây xác định ASAH1 là gen nhạy cảm với sẹo lồi có tính gia đình. Thông qua phân tích dữ liệu toàn bộ bộ gen, một locus trên 8p23.3-p21.3 được lập bản đồ với điểm LOD là 4,48. Giải trình tự toàn bộ exome tập trung vào locus này sau đó xác định được đột biến sai nghĩa c.1202T>C (p.Leu401Pro) trong gen Nacylsphingosine amidohydrolase (ASAH1) trong một họ Yoruba lớn. Gen ASAH1 mã hóa cho enzyme “acid ceramidase”, chịu trách nhiệm phân hủy ceramide. Ceramide liên quan đến quá trình hình thành khối u và sự gia tăng ceramide nội bào có thể gây ra tín hiệu chết theo chương trình [68]. Protein ASAH1 được biểu hiện nhiều trong các dòng tế bào ung thư và đại thực bào phế nang ở người, cho thấy vai trò trong các tổn thương tăng sinh và viêm. Mặc dù kiểu gen của ASAH1 đồng phân ly với kiểu hình sẹo lồi trong họ lớn, nhưng mô sẹo lồi không được lấy từ những người mang biến thể ASAH1. Do đó, hồ sơ bệnh lý và mức độ ceramide và ceramidase trong sẹo lồi biểu hiện biến thể này có thể được xác minh. Cần có thêm các nghiên cứu chức năng để làm rõ vai trò của biến thể ASAH1 trong quá trình sinh bệnh sẹo lồi.
4.7 Triển vọng trong tương lai
RNA-seq (giải trình tự RNA), còn được gọi là giải trình tự shotgun toàn bộ phiên mã (WTSS), là một công nghệ cũng sử dụng khả năng của giải trình tự thế hệ tiếp theo để tiết lộ ảnh chụp nhanh về sự hiện diện và số lượng RNA từ bộ gen tại một thời điểm nhất định. Do đó, dữ liệu RNA-seq hữu ích trong việc giải thích “phiên mã cá nhân hóa” và có thể được áp dụng để cải thiện sự hiểu biết về những thay đổi trong hồ sơ phiên mã trong quá trình sinh học sẹo lồi. Onoufriadis và cộng sự áp dụng RNA-seq để khám phá những thay đổi phiên mã tại một thời điểm sớm hơn—trong quá trình hình thành sẹo lồi. Tiến hành sinh thiết-tái sinh thiết mông của tám đối tượng dễ bị sẹo lồi và sáu đối chứng khỏe mạnh sau khoảng thời gian 6 tuần. So sánh đối chứng khỏe mạnh trước và sau khi bị thương xác định được 2215 gen biểu hiện khác biệt, trong khi cùng một phân tích ở những cá nhân dễ bị sẹo lồi xác định được 3161 gen biểu hiện khác biệt. Trong số những gen đó, có 513 gen đặc hiệu cho những cá nhân khỏe mạnh và 1449 gen đặc hiệu cho kiểu hình sẹo lồi. Có 22 con đường liên quan trực tiếp đến những cá nhân dễ bị sẹo lồi. Lưu ý, tín hiệu NOTCH, tín hiệu MAPK và các con đường thụ thể giống Toll được phát hiện là bị thay đổi ở những cá nhân dễ bị sẹo lồi sau khi bị thương. Phân tích mạng lưới liên kết gen chứng minh được thông tin biểu hiện trung bình khác nhau của các gen truyền tín hiệu cytokine giữa những cá nhân dễ bị sẹo lồi và những cá nhân khỏe mạnh trong quá trình lành thương. Nghiên cứu cung cấp một phân tích toàn diện và tích hợp về bản sao mã sẹo lồi và làm nổi bật các con đường sinh học đặc trưng trong quá trình hình thành sẹo lồi. Quan trọng hơn, kết quả có thể giúp xác định các yếu tố thúc đẩy gen.
4.8 Kết luận
Mặc dù có nhiều bằng chứng hoặc hiện tượng cho thấy thành phần di truyền trong quá trình hình thành sẹo lồi, vẫn còn rất nhiều công việc cần thực hiện để tăng cường hiểu biết hiện tại của chúng ta về cơ sở di truyền của quá trình hình thành sẹo lồi. Các nghiên cứu trước đây gợi ý khả năng có mô hình di truyền đa gen trong sẹo lồi. Ngoài ra, nhiều gen hoặc vị trí gen được xác định trong quá trình bệnh sinh của sẹo lồi. Tuy nhiên, hầu hết các phát hiện này đều cần các nghiên cứu về chức năng sâu hơn để làm rõ khả năng gây bệnh. Hy vọng rằng những nghiên cứu như vậy sẽ giúp làm sáng tỏ cơ sở phân tử của quá trình hình thành sẹo lồi và đưa ra các chiến lược mới để phòng ngừa, chẩn đoán và điều trị.
Tài liệu tham khảo
- Tuan TL, Nichter LS. The molecular basis of keloid and hypertrophic scar formation. Mol Med Today. 1998;4(1):19–24.
- Lee JY, Yang CC, Chao SC, Wong TW. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26(5):379–84.
- Brissett AE, Sherris DA. Scar contractures, hypertrophic scars, and keloids. Facial Plast Surg. 2001;17(4):263–72.
- Robles DT, Moore E, Draznin M, Berg D. Keloids: pathophysiology and management. Dermatol Online J. 2007;13(3):9.
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314–21.
- Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med. 2011;17(1–2):113– 25.
- Al-Attar A, Mess S, Thomassen JM, Kauffman CL, Davison SP. Keloid pathogenesis and treatment. Plast Reconstr Surg. 2006;117(1):286–300.
- Butler PD, Longaker MT, Yang GP. Current progress in keloid research and treatment. J Am Coll Surg. 2008;206(4):731–41.
- He Y, Deng Z, Alghamdi M, Lu L, Fear MW, He L. From genetics to epigenetics: new insights into keloid scarring. Cell Prolif. 2017;50:e12326.
- Huang C, Murphy GF, Akaishi S, Ogawa R. Keloids and hypertrophic scars: update and future directions. Plast Reconstr Surg Glob Open. 2013;1(4):e25.
- Berman B, Maderal A, Raphael B. Keloids and hypertrophic scars: pathophysiology, classification, and treatment. Dermatol Surg. 2017;43(Suppl 1):S3–S18.
- Hsu CK, Lin HH, Harn HI, Hughes MW, Tang MJ, Yang CC. Mechanical forces in skin disorders. J Dermatol Sci. 2018;90(3):232–40.
- Ogawa R, Hsu CK. Mechanobiological dysregulation of the epidermis and dermis in skin disorders and in degeneration. J Cell Mol Med. 2013; 17(7):817–22.
- Glass DA 2nd. current understanding of the genetic causes of keloid formation. J Investig Dermatol Symp Proc. 2017;18(2):S50–S3.
- Kelly AP. Keloids. Dermatol Clin. 1988; 6(3): 413–24.
- LeFlore IC. Misconceptions regarding elective plastic surgery in the black patient. J Natl Med Assoc. 1980;72(10):947–8.
- Ud-Din S, Bayat A. Strategic management of keloid disease in ethnic skin: a structured approach supported by the emerging literature. Br J Dermatol. 2013;169(Suppl 3):71–81.
- Child FJ, Fuller LC, Higgins EM, Du Vivier AW. A study of the spectrum of skin disease occurring in a black population in southeast London. Br J Dermatol. 1999;141(3):512–7.
- Marneros AG, Norris JE, Olsen BR, Reichenberger E. Clinical genetics of familial keloids. Arch Dermatol. 2001;137(11):1429–34.
- Omo-Dare P. Genetic studies on keloid. J Natl Med Assoc. 1975;67(6):428–32.
- Chen Y, Gao JH, Liu XJ, Yan X, Song M. Characteristics of occurrence for Han Chinese familial keloids. Burns. 2006;32(8):1052–9.
- Clark JA, Turner ML, Howard L, Stanescu H, Kleta R, Kopp JB. Description of familial keloids in five pedigrees: evidence for autosomal dominant inheritance and phenotypic heterogeneity. BMC Dermatol. 2009;9:8.
- Phillips DI. Twin studies in medical research: can they tell us whether diseases are genetically determined? Lancet. 1993;341(8851):1008–9.
- Bloom D. Multiple keloids in twin sisters. Arch Derm Syphilol. 1947;55(3):426.
- Siraganian PA, Rubinstein JH, Miller RW. Keloids and neoplasms in the Rubinstein-Taybi syndrome. Med Pediatr Oncol. 1989;17(6):485–.
- van de Kar AL, Houge G, Shaw AC, de Jong D, van Belzen MJ, Peters DJ, et al. Keloids in Rubinstein-Taybi syndrome: a clinical study. Br J Dermatol. 2014;171(3):615–21.
- Warner DR, Bhattacherjee V, Yin X, Singh S, Mukhopadhyay P, Pisano MM, et al. Functional interaction between Smad, CREB binding protein, and p68 RNA helicase. Biochem Biophys Res Commun. 2004;324(1):70–6.
- Korzus E. Rubinstein-Taybi syndrome and epigenetic alterations. Adv Exp Med Biol. 2017;978:39–62.
- Diao JS, Xia WS, Yi CG, Wang YM, Li B, Xia W, et al. Trichostatin A inhibits collagen synthesis and induces apoptosis in keloid fibroblasts. Arch Dermatol Res. 2011;303(8):573–80.
30. - normal and keloid scars. J Invest Dermatol. 2012;132(4):1293–6.
- Russell SB, Russell JD, Trupin KM, Gayden AE, Opalenik SR, Nanney LB, et al. Epigenetically altered wound healing in keloid fibroblasts. J Invest Dermatol. 2010;130(10):2489–96.
- Atwal PS, Blease S, Braxton A, Graves J, He W, Person R, et al. Novel X-linked syndrome of cardiac valvulopathy, keloid scarring, and reduced joint mobility due to filamin A substitution G1576R. Am J Med Genet A. 2016;170a(4):891– 5.
- Sasaki A, Masuda Y, Ohta Y, Ikeda K, Watanabe K. Filamin associates with Smads and regulates transforming growth factor-beta signaling. J Biol Chem. 2001;276(21):17871–7.
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8– 26.
- Burk CJ, Aber C, Connelly EA. Ehlers-Danlos syndrome type IV: keloidal plaques of the lower extremities, amniotic band limb deformity, and a new mutation. J Am Acad Dermatol. 2007;56(2 Suppl):S53–4.
- Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11(3):241–7.
- Marneros AG, Norris JE, Watanabe S, Reichenberger E, Olsen BR. Genome scans provide evidence for keloid susceptibility loci on chromosomes 2q23 and 7p11. J Invest Dermatol. 2004;122(5):1126–32.
- Yan X, Gao JH, Chen Y, Song M, Liu XJ. Preliminary linkage analysis and mapping of keloid susceptibility locus in a Chinese pedigree. Zhonghua Zheng Xing Wai Ke Za Zhi. 2007;23(1):32–5.
- Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363(2):166–76.
- Nakashima M, Chung S, Takahashi A, Kamatani N, Kawaguchi T, Tsunoda T, et al. A genome-wide association study identifies four susceptibility loci for keloid in the Japanese population. Nat Genet. 2010;42(9):768–71.
- Chung S, Nakashima M, Zembutsu H, Nakamura Y. Possible involvement of NEDD4 in keloid formation; its critical role in fibroblast proliferation and collagen production. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87(8):563–73.
- Vecchione A, Marchese A, Henry P, Rotin D, Morrione A. The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Mol Cell Biol. 2003;23(9):3363–72.
- Daian T, Ohtsuru A, Rogounovitch T, Ishihara H, Hirano A, Akiyama-Uchida Y, et al. Insulin-like growth factor-I enhances transforming growth factor-beta-induced extracellular matrix protein production through the P38/activating transcription factor-2 signaling pathway in keloid fibroblasts. J Invest Dermatol. 2003;120(6):956– 62.
- Ishihara H, Yoshimoto H, Fujioka M, Murakami R, Hirano A, Fujii T, et al. Keloid fibroblasts resist ceramide-induced apoptosis by overexpression of insulin-like growth factor I receptor. J Invest Dermatol. 2000;115(6):1065– 71.
- Yoshimoto H, Ishihara H, Ohtsuru A, Akino K, Murakami R, Kuroda H, et al. Overexpression of insulin-like growth factor-1 (IGF-I) receptor and the invasiveness of cultured keloid fibroblasts. Am J Pathol. 1999;154(3):883–9.
- Zhu F, Wu B, Li P, Wang J, Tang H, Liu Y, et al. Association study confirmed susceptibility loci with keloid in the Chinese Han population. PLoS One. 2013;8(5):e62377.
- Ogawa R, Watanabe A, Than Naing B, Sasaki M, Fujita A, Akaishi S, et al. Associations between keloid severity and single-nucleotide polymorphisms: importance of rs8032158 as a biomarker of keloid severity. J Invest Dermatol. 2014;134(7):2041–3.
- Shriner D. Overview of admixture mapping. Curr Protoc Hum Genet. 2013;Chapter 1:Unit 1.23.
- Velez Edwards DR, Tsosie KS, Williams SM, Edwards TL, Russell SB. Admixture mapping identifies a locus at 15q21.2-22.3 associated with keloid formation in African Americans. Hum Genet. 2014;133(12):1513–23.
- Hsu CK, Lin HH, Harn HI, Ogawa R, Wang YK, Ho YT, et al. Caveolin-1 controls hyperresponsiveness to mechanical stimuli andfibrogenesis-associated RUNX2 activation in keloid fibroblasts. J Invest Dermatol. 2018;138(1):208–18.
- Harn HI, Hsu CK, Wang YK, Huang YW, Chiu WT, Lin HH, et al. Spatial distribution of filament elasticity determines the migratory behaviors of a cell. Cell Adh Migr. 2016;10(4):368–77.
- Harn HI, Wang YK, Hsu CK, Ho YT, Huang YW, Chiu WT, et al. Mechanical coupling of cytoskeletal elasticity and force generation is crucial for understanding the migrating nature of keloid fibroblasts. Exp Dermatol. 2015;24(8):579–84.
- Shih B, Bayat A. Comparative genomic hybridisation analysis of keloid tissue in Caucasians suggests possible involvement of HLA-DRB5 in disease pathogenesis. Arch Dermatol Res. 2012;304(3):241–9.
- Shih B, Bayat A. Genetics of keloid scarring. Arch Dermatol Res. 2010;302(5):319–39.
- Brown JJ, Bayat A. Genetic susceptibility to raised dermal scarring. Br J Dermatol. 2009;161(1):8–18.
- Brown JJ, Ollier WE, Thomson W, Bayat A. Positive association of HLA-DRB1∗15 with keloid disease in Caucasians. Int J Immunogenet. 2008;35(4-5):303–7.
- Brown JJ, Ollier W, Arscott G, Ke X, Lamb J, Day P, et al. Genetic susceptibility to keloid scarring: SMAD gene SNP frequencies in AfroCaribbeans. Exp Dermatol. 2008;17(7):610–3.
- He S, Liu X, Yang Y, Huang W, Xu S, Yang S, et al. Mechanisms of transforming growth factor beta(1)/Smad signalling mediated by mitogenactivated protein kinase pathways in keloid fibroblasts. Br J Dermatol. 2010;162(3):538–46.
- Emami A, Halim AS, Salahshourifar I, Yussof SJ, Khoo TL, Kannan TP. Association of TGFbeta1 and SMAD4 variants in the etiology of keloid scar in the Malay population. Arch Dermatol Res. 2012;304(7):541–7.
- Bayat A, Bock O, Mrowietz U, Ollier WE, Ferguson MW. Genetic susceptibility to keloid disease and transforming growth factor beta 2 polymorphisms. Br J Plast Surg. 2002;55(4):283–6.
- Bayat A, Bock O, Mrowietz U, Ollier WE, Ferguson MW. Genetic susceptibility to keloid disease and hypertrophic scarring: transforming growth factor beta1 common polymorphisms and plasma levels. Plast Reconstr Surg. 2003;111(2):535–43. discussion 44-6.
- Bayat A, Bock O, Mrowietz U, Ollier WE, Ferguson MW. Genetic susceptibility to keloid disease: transforming growth factor beta receptor gene polymorphisms are not associated with keloid disease. Exp Dermatol. 2004;13(2):120–4.
- Bayat A, Walter JM, Bock O, Mrowietz U, Ollier WE, Ferguson MW. Genetic susceptibility to keloid disease: mutation screening of the TGFbeta3 gene. Br J Plast Surg. 2005;58(7):914–21.
- Tu Y, Lineaweaver WC, Zhang F. TGF-beta1- 509C/T polymorphism and susceptibility to keloid disease: a systematic review and metaanalysis. Scars Burn Heal. 2017;3:2059513117709943.
- Saed GM, Ladin D, Olson J, Han X, Hou Z, Fivenson D. Analysis of p53 gene mutations in keloids using polymerase chain reaction- based single-strand conformational polymorphism and DNA sequencing. Arch Dermatol. 1998;134(8):963–7.
- Yan L, Lu XY, Wang CM, Cao R, Yin YH, Jia CS, et al. Association between p53 gene codon 72 polymorphism and keloid in Chinese population. Zhonghua Zheng Xing Wai Ke Za Zhi. 2007;23(5):428–30.
- Santos-Cortez RLP, Hu Y, Sun F, BenahmedMiniuk F, Tao J, Kanaujiya JK, et al. Identification of ASAH1 as a susceptibility gene for familial keloids. Eur J Hum Genet. 2017;25(10):1155–61.
- Huang WC, Chen CL, Lin YS, Lin CF. Apoptotic sphingolipid ceramide in cancer therapy. J Lipids. 2011;2011:565316.
- Coant N, Sakamoto W, Mao C, Hannun YA. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv Biol Regul. 2017;63:122–31.
- Chu Y, Corey DR. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic Acid Ther. 2012;22(4):271–4.
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63.
- Onoufriadis A, Hsu CK, Ainali C, Ung CY, Rashidghamat E, Yang HS, et al. Time series integrative analysis of RNA sequencing and microRNA expression data reveals key biologic wound healing pathways in keloid-prone individuals. J Invest Dermatol. 2018;138:2690–.
- Zhang G, Jiang J, Luo S, Tang S, Liang J, Yao P. Analyses of CDC2L1 gene mutations in keloid tissue. Clin Exp Dermatol. 2012;37(3):277–83.